
What does it take to turn a botanical into an FDA-Approved drug?
The FDA has a unique, streamlined process for medicinally active plant-based products to gain market authorization. That this process is much faster...
3 min read
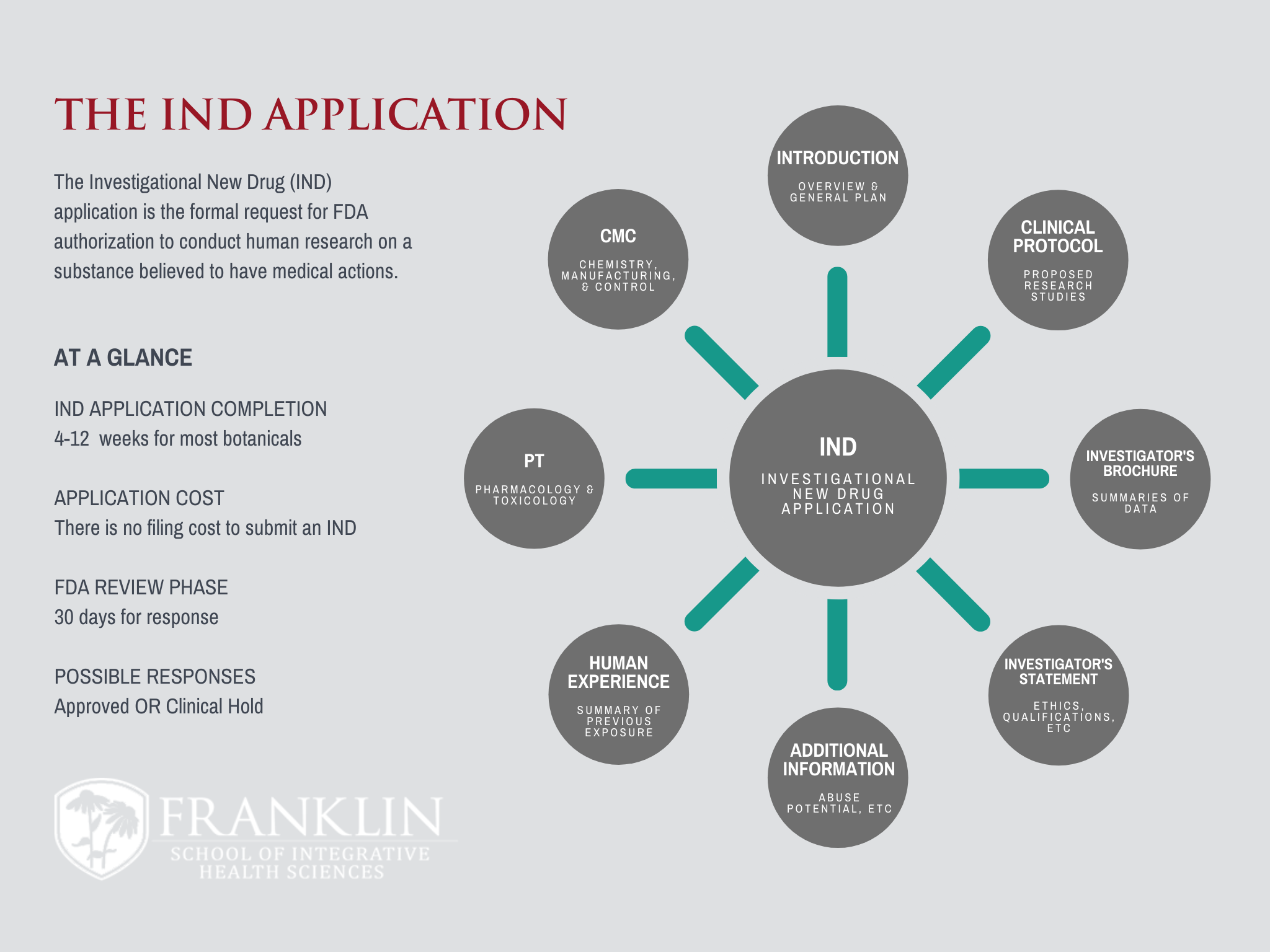

The FDA has a unique, streamlined process for medicinally active plant-based products to gain market authorization. That this process is much faster...

Outcome measures are among the most critical decisions in clinical trial design. After all, this is what determines whether or not your product was...
